和评论:化学》杂志上的雷竞技苹果下载研究表明
e-ISSN: 2319 - 9849
e-ISSN: 2319 - 9849
1维特大学化学系钦奈校园,钦奈127年- 600年,印度泰米尔纳德邦,
2化学系,阿卜杜勒·卡里姆·阿里Shayad工学院和理工,MMANTC, Mansoora,马勒冈203年- 423年,印度马哈拉施特拉邦
收到日期:25/04/2018;接受日期:02/05/2018;发表日期:18/05/2018
访问更多的相关文章dota2雷竞技
苯并[1,2 b: 6、5 b '] dithiophene-4 5-dione (BDTD)作为一个中间合成受体单元用于有机太阳能电池。但它有双供体和受体的活动还没有测试。在这项研究中,BDTD, DTBT DTBTD视为捐赠者和确定其光电特性以及benzothiadiazole一半。带隙、HOMO LUMO和吸收波长时审查和研究光电性质如何影响引入吸电子替换如F, CN和NO2在benzothiadiazole单位。间的距离和真正的电子供体和受体系统识别在电子从基态和激发态的计算密度。强烈的氢键供体和受体之间的交互和强烈的空间排斥单位观察而引入不同的取代基电子撤回到benzothiadiazole受体单元。片段的贡献在分子轨道能级支持DTBT的供者特征的结果,DTBTD BDTD和双重性格。新设计的分子到达电力转换效率我´scharber图预测的8%。
密度泛函理论,改变密度泛函理论,HOMO和LUMO, Scharber,非共价相互作用,Benzothiadiazole
过去二十年有机低带隙共聚物有几个优势在有机太阳能电池等电子设备,OLED的1,2],OFET [3),内存芯片(4),化学传感器(5),有机非线性光学材料等由于低成本的潜在应用6)、快速和廉价的轧辊辊生产和低体重7),使其替代无机光电设备。电源转换效率(PCE)的聚合物太阳能电池增强迅速从低于0.1%到13%在过去的二十年里8,9]。这令人印象深刻的成就主要是通过共轭聚合物的结构设计与战略不同的捐赠者和受体。最近,母鸡等人报道了有机太阳能电池的能量转换效率11.5%是高排名的设备到目前为止在有机光伏电池的线10]。口服脊髓灰质炎疫苗,共轭骨干直接共轭聚合物的重要组成部分,因为它说,共轭聚合物的物理性质如带隙、原子的相互作用,不同的取代基(N, O,年代,Se…)也调整共轭聚合物的物理性质和侧链是重要的工具来提高聚合物的溶解度(11- - - - - -13]。在有机共轭聚合物,平面几何提高有效共轭链的长度和降低HOMO和LUMO能级之间的带隙,与平面结构可以提高远程批量邻国之间的分子间的相互作用,堆放p-conjugated骨干(14]。明确,我们相信Vinylene-linked共轭聚合物可以进行一系列的相关优势更传统的系统,包括整平相邻芳单位和ethenylic间距器(15],扩展p-conjugation [16),高效的离域(17),因此,减少了带隙和增强的光学性质在低能光谱区(18,19]。噻吩融合材料的稳定性较高的线性相关结构,与更有效的结合并相互也表现出各种内部和分子间的相互作用,如范德华相互作用,弱氢键,p p堆积往往导致高电荷流动[20.- - - - - -24]。最近,标志等人报道BDTD由材料2、7-bis ((5-perfluorophenacyl) thiophen-2-yl) 9日10-phenanthrenequinone;2、7-bis ((5 - phenacyl) thiophen-2-yl) 9日10-phenanthrenequinone;和2,7-bis (thiophen-2-yl) 9日10-phenanthrenequinone最大吸收范围的369,370,和318 nm分别明显蓝移,他们使用作为一个受体单元BDTD一起捐赠噻吩单元和不同取代基对其拥有的带隙的大小为2.62,2.52和2.56 eV,分别得到了明显的人类价值超过6.5 eV由于两个羰基存在(25]。相反,我们使用BDTD单位作为供体,研究其电子捐赠贡献倾向而混合BTD用作受体。因为一部分BDTD本身具有双重到和两个由于吐温的存在p过多bi-thiophene和强劲的电子撤回酮组(26]。最近,小弗朗哥等人报道各种电子捐赠(- nh2 -哦,…等)和撤回取代基(如F, CN、NO2、CF3)显著改变HOMO和LUMO能级(27),由于能量分布不均(不平等系数)的,因为更高的电负性的大小比碳(氮和氧28]。现在,我们有一个非常强大的和相对低成本应用预测有机和无机材料的光电特性在某种程度上通过量子力学计算。新设计的光电特性取得了共聚物通过引入DFT和TD-DFT计算差异的带隙HOMO和LUMO激发能级和B3LYP / 6 - 31 g * *基础设置,尽管这可能让位于p共轭的实质性的错误扩展系统和远程充电(CT)转移励磁系统(29日- - - - - -31日]。然而,TDDFT采用传统exchange-correlation功能提供了相当令人印象深刻的轨道能级和价电子激发能量分别为有机和无机材料(32]。在本文中,我们把我们的注意力放在benzothiadiazole与三个不同的捐赠单位如苯并[1,2 b: 6、5 b '] dithiophene-4 5-dione (BDTD);(3 ',2 ':3、4、2”,3”:5、6]苯并[1,2 - d] [1、2、3-triazole (DTBT);Thieno[3“2”: 3、4、2”, 3”: 5、6]苯并(2 c) (DTBTD),介绍了一种新型润滑脂添加剂噻二唑衍生物三个不同的电子撤回取代基是F, CN和NO2受体单元,研究了带隙,吸收有条不紊地调谐共轭单体单位使用量子力学计算DFT和TDDFT方法。在这项研究中,我们探讨了影响电子撤回组到benzothiadiazole单元与三个不同的BDTD DTBT,和DTBTD根几何、电子结构、电荷转移长度、电荷密度差异,电子空穴分布和非共价相互作用。理论上预测光电性能的单体和二聚体模型代表聚合物的性质(33,34]。因此,我们通过密度泛函理论方法研究了光电性质十二个新设计的单体半个。在这项研究中我们已经讨论了(我)字符DTBT, DTBTD和BDTD DFT方法截然不同的研究电荷密度差异,电荷转移方向和电荷转移距离在电子跃迁和片段分子轨道能级和贡献(ii)取代基电子撤回如何改变分子的HOMO和LUMO能级和几何。
几何优化和仿真的吸收光谱进行了密度泛函和时间功能理论进行了高斯09年软件包(35]。每个单元的几何优化进行了分子在基态的B3LYP /我感觉*基础设置配置在气相没有任何环约束与限制整个系统电子自旋为了得到精确值,因此计算成本控制。基态能级和激发态能级基于DFT和TDDFT计算,B3LYP和我(d, p)基组来确定地面(S0)和激发态能级(Sn)。眼压(9/40 = 4)关键字用于TDDFT计算为了得到更多的配置系数在同一水平上。电子空穴分布、共价相互作用(NCI),电荷转移(长度36CT (? r)和电荷密度差异(CDD)地图已被用于电子过渡期间仔细观察分子内电荷转移实现Multiwfn [37软件套件。共价相互作用的计算进行wB97XD / 6 - 311 g (d, p)水平的DFT。wB97XD功能的非共价相互作用的可靠的方法,因为比B3LYP准确性,B2PLYP-D3 B3LYP-D3方法也避免的价钱(B3LYP)和过度弯曲(B3LYPD) [38- - - - - -40]。电子空穴函数被使用Gabedit软件包[策划41]。NCI研究化合物的表面通过使用视觉模拟分子动力学(42软件套件。
十二个不同的单体的基态几何优化的DFT-B3LYP /我感觉*代替DFTB3LYP / 3-21G *, M1-M12优化几何图形所示图1 b。非共价相互作用研究是理解分子内弱相互作用中发挥作用的调优共轭体系的能级变化强烈的电子撤回替换到benzothiadiazole单元和几何的共轭体系。优化几何图形的三硝基取代单体M4, M8, M12和二面角角度所示图1 c。二面角(?)参数共轭体系的骨干单位之间有显著影响的共轭聚合物,因此允许改变光电性质。取代基的立体互动到供体/受体单元和共轭骨干应该避免在设计一个共轭系统,因为导致减少有效共轭长度结果指导的紫外可见吸收蓝移(43,44]。未被取代的优化结构,F和腈(CN)组替换到benzothiadiazole单位观察与DTBT拥有完美的平面,BDTD DTBTD单位,没有观察到相当大的扭转角。另一方面单体半个M4, M8, M12观察平面化的最大偏离M4的顺序(15°/ 6°)< M8 (15°/ 24°) < M12 (24°/ 25°)。在M4, M8和M14硝基氧原子都表现出的飞机。一个分子间p - p重叠和共轭体系的电荷传输与刚性结构可以通过平面配置整个分子的共轭骨干(p -45]。系统M1-M3、M5-M7 M9-M11十二观察完全平面中九个系统配置,可以解释为非共价相互作用分析(NCI)是基于密度梯度下降之间的关系和电子密度?(r)。NCI的等值面分析允许降低在最低密度梯度图像的位置和性质在三维空间中《海军罪案调查处》。NCI分析由wB97XD / 6 - 311 g (d, p)水平的DFT克服目标理论的局限性(46]。NCI的等值面分子阴谋所示图2彩色地图识别类型的交互。对所有系统,h氢键相互作用是观察BTD单元和2 -亚乙烯基之间的联系,在氢键表示蓝色的颜色NCI等值面图。F和CN替换系统有氢键之间的(N - h和F - h)乙烯质子和BTD腈和硝基氮和F替换到BTD单位。这显然表明,扭转角度消除p-extended系统之间的氢键的烯键和负责任地BTD单位领导高度平面p -扩展系统的烯键和BTD单位负责领导高度平面p-conjugated系统。观察到三个系统(M4, M8和M12)展出non-planar几何空间斥力的oo的两个硝基BTD上。很明显从我们的研究证据,确认会解锁通过引入两个硝基组到BTD单位。通过分子的几何关联其带隙能理解分子的几何形状影响带隙。九个分子展出M1-M3平面几何,M5-M7, M9-M11及其带隙是2.17,2.15,1.91,2.21,2.17,1.96,1.96,2.23和2.02 eV。硝基取代分子M4, M8和M12展出non-planar几何和带隙是1.85,1.85和2.07 eV。宽的带隙M12认为由于存在两个强大的电子撤回硝基和两个酮残留在分子的HOMO能级比LUMO能级的不稳定。 This above results from our study showed that planarity of the geometry strongly affected the band gap of the conjugated system.

图1:答:设计的化合物的化学结构;b .优化几何;c .二面角和债券长度的分子M4, M8和M12。

图2:NCI iso-surface和情节的密度梯度和研究分子的电子密度。
HOMO, LUMO和共轭系统的带隙能级直接告诉我们代表聚合物的光电性能。所有优化单体带隙HOMO和LUMO能级之间的能量差在B3LYP /我感觉(d, p)基组。在这里,我们使用单体找出代表的带隙能级系统中,为了设计非常狭窄的带隙聚合物。人类,LUMO和带隙值研究了分子中列出表1。前沿分子轨道的单位和研究单体显示在半个分开图3 b。苯并,naphtho和人类学融合噻吩环的共轭系统给出了较低的带隙融合戒指由LUMO能级稳定和不稳定的HOMO芳香形式反之亦然的醌型的形式(47,48]。分离供体和受体进行了几何优化和能量水平的B3LYP /我感觉DFT (d, P)方法。之前报道的结果,DTBT和DTBTD单位作为电子受体在捐赠系统由于存在五元电子缺陷环(20.,49,50]。相比之下,DTBT DTBTD单位我们被认为是和作为电子供体,因为两个单位自己有双供体和受体的活动将同时拥有p过多bithiophene和电子撤回/不足三唑DTBT和BDTD DTBTD像一种新型润滑脂添加剂噻二唑衍生物。DTBT拥有高——撒谎比DTBTD HOMO和LUMO能级;而BTD有低洼比DTBT和DTBTD LUMO能级。这个结果表明BTD强烈倾向于接受一个电子从DTBT和DTBTD。而F、CN和NO2代替BTD显示发生在深低洼LUMO能级。硝基取代受体DNBTD LUMO能级的-3.70 eV比在所有深度降低。以上结果表明,电子撤回组到共轭系统的影响大大动摇LUMO能级。受体的LUMO能级下降的顺序BTD > DFBTD > DCNBTD > DNBTD。所以DNBTD是比它更强的受体前体,它倾向于降低共轭聚合物的带隙。 The band gap (Eg) of M3, M4, M7, M8, and M9 are 1.91, 1.85, 1.96, 1.85, 1.96 eV respectively. These values were observed band gap less than 2.0 eV. By contrast, band gaps of M11 and M12 are 2.02 and 2.07 eV respectively, due to deep lowered HOMO energy levels than LUMO band gap and low energy level of HOMO may possess high open circuit voltage when applied to bulk heterojunction solar cells. It was observed that LUMO energy levels were destabilised for all systems by electron withdrawing substituent’s in the order of F>CN>NO2. For M1-M4 and M5-M8 monomers, N=O---H-C, CN---H-C and F---H-C hydrogen bond interaction was much higher than Unsubstituted systems resulting in lower the band gap of the system. From the molecular orbital spatial distribution (图3 d)的分子M1-M12,人类主要是局部的密度DTBT DTBTD单位,而BTD LUMO主要是局部的,DFBTD DCNBTD, DNBTD单位。清楚表明DTBT和DTBTD电子捐赠趋势比受体趋势报告的其他研究小组20.,50]。相比之下,在M9-M12 HOMO和LUMO同样BDTD本地化,BTD DFBTD DCNBTD, DNBTD单位。的明确证据和理解字符DTBT, BDTD DTBTD单位,碎片的贡献在分子轨道能级进行了利用chemissian计划。
| 化合物 | EHOMO (eV) | ELUMO (eV) | 吗?LUMO PC61BM (eV) | 吗?LUMO ITIC-Th1 (eV) | Egap (eV) | 吗?马克斯(nm) | f | 吗?一个 | Voc (V) |
| M1 | -5.00 | -2.83 | 1.47 | 1.18 | 2.17 | 643年 | 1.2769 | 0.9471 | 0.40 (0.69) |
| 平方米 | -5.11 | -2.96 | 1.34 | 1.05 | 2.15 | 651年 | 1.2077 | 0.9380 | 0.51 (0.8) |
| M3 | -5.36 | -3.45 | 0.85 | 0.56 | 1.91 | 735年 | 1.1219 | 0.9244 | 0.76 (1.05) |
| M4 | -5.41 | -3.56 | 0.74 | 0.45 | 1.85 | 776年 | 0.8230 | 0.8496 | 0.81 (1.1) |
| M5 | -5.17 | -2.96 | 1.34 | 1.05 | 2.21 | 636年 | 1.3982 | 0.9600 | 0.57 (0.86) |
| M6 | -5.27 | -3.10 | 1.3 | 0.91 | 2.17 | 642年 | 1.3195 | 0.9520 | 0.67 (0.96) |
| M7 | -5.52 | -3.56 | 0.74 | 0.45 | 1.96 | 720年 | 1.1771 | 0.9334 | 0.92 (1.21) |
| M8 | -5.57 | -3.72 | 0.58 | 0.29 | 1.85 | 783年 | 0.7815 | 0.8346 | 0.97 (1.26) |
| M9 | -5.44 | -3.48 | 0.82 | 0.53 | 1.96 | 760年 | 0.5666 | 0.7287 | 0.84 (1.13) |
| M10 | -5.60 | -3.37 | 0.93 | 0.64 | 2.23 | 652年 | 0.7387 | 0.8174 | 1.00 (1.29) |
| M11公路 | -5.82 | -3.80 | 0.47 | 0.18 | 2.02 | 711年 | 1.1445 | 0.9283 | 1.22 (1.51) |
| M12 | -5.90 | -3.83 | 0.47 | 0.18 | 2.07 | 708年 | 0.9540 | 0.8888 | 1.30 (1.59) |
| ITIC-Th1 [74] | -5.74 | -4.01 | - - - - - - | 1.73 | 728年 | - - - - - - | - - - - - - | - - - - - - | |
| PC61BM [75] | -6.00 | -4.3 | - - - - - - | - - - - - - | - - - - - - | - - - - - - | - - - - - - | - - - - - - |
表1。计算HOMO、LUMO ? LUMO (PC61BM),和? LUMO (ITIC-Th1), UV / vis吸收(nm),振子强度,光吸收效率(?)和开路电压(V) (PCBM LUMO能级是用来确定Voc的研究分子;值与ITIC-Th1括号。

图3:fmo的独立的单位;b。前沿分子轨道能量为调查十二分子6-31 G (d, p)水平的理论;c。模拟吸收光谱和d。分子轨道空间分布。
吸收光谱、振子强度和电荷转移的长度
光吸收范围和强度在短路电流中发挥至关重要的作用(Jsc)光伏设备。所有的单体都计算在TD-DFT-B3LYP /我感觉* *水平。模拟吸收光谱,振子强度(ƒ)和十二个新设计的光吸收共轭单体中列出表1。光吸收效率(?)表示为:
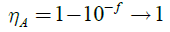
在这里。ƒ的振子强度(强度)是代表吸收波长和广泛的光吸收共轭聚合物导致减少所需厚度的光吸收材料通过优化电压生成、填充因子和改善自由电荷载体(51- - - - - -54]。在含杂原子共轭有机分子发挥重要作用使吸收红移或蓝移。现在的渴求增加singlet-singlet [S] _ (0) ? S_1) n ?p ^ *过渡到通过培养对近红外光谱(近红外)地区可能是由于激发的非键电子从分子轨道(通常是杂原子)p分子平面(55)和红移可以解释为持续p *激发态(56)和阻燃剂*、p - p *债券红移与p共轭系统当羰基共轭。57Now a day’s hunt to get singlet-singlet transition to passage of cultivation towards NIR region due to excitation of non-bonding electrons (usually hetero atoms) from molecular orbital to p molecular plane [57- - - - - -63年),红移可以解释为p *激发态的坚持64年n]也?p *和?p *带红移当共轭羰基与p共轭系统(65年- - - - - -67年]。吸收波长的M1, M2, M5, M6 M10和643年,651年,636年、642年和652年分别纳米所示图3 c。这是研究化合物单体中观察到M5举行非常低的吸收波长636 nm。表1中列出,光吸收效率来源于振子强度明显表明,充足的光吸收效率观察到所有单位。因此,“协同效应”伴随着氢键相互作用导致产生共轭系统和狭窄的带隙宽吸收波长特性。电子的转移从供体受体称为形成(D + -) *激发态的过程强烈取决于不同类型的化学受体和捐赠者之间的链接/单元单位。电荷密度的区别、电子转移的方向和电荷转移距离r(?)被TD-DFT计算方法对一个家庭的新设计的推挽式分子。电荷转移的距离、方向和电荷密度差分的计算是通过报道方法(36,68年]。电子跃迁和相应的参数表1中列出,显示的电子跃迁特征最大吸收转换(S0 ? S1)的单体都是相同的。的电子密度分布的homo和lumo分子是由分子本身的电荷转移(副69年]。电子转换涉及HOMO和LUMO轨道是分子内电荷转移激发态,电子从电子供体的电子受体单元。电荷密度的不同情节(电子空穴分布)所示图4。从电子密度图单体M1-M8洞(浅绿色)本地化几乎在benzothiadiazole电子本地化DTBT和DTBTD单位。相比之下,在单体M9和M10 lumo轨道的电子分布BDTD单位而不是BTD本地化,而在单体M11公路和M12 lumo是本地化的电子分布主要在DCNBTD和DNBTD单位。费用和电荷转移的重心长度和分子轨道(MO)贡献为了证明电子密度,在电子电荷转移方向过渡。从分析电荷转移方向绘制图4一,这显然是显示大量的重叠指控代表区域的质心的放大,对激发电子密度的降低。电荷转移计算当从M1 M4展品洞DTBT单元,在一个电子BTD-DNBTD单位用轨道的(蓝洞;greenelectron)。M5-M8系统类似的结果观察,这清楚地表明一个电子转移从DTBT单元BTD和EWG代替BTD M5-M8也。这显然是暗示DTBT DTBTD也倾向于贡献一个电子电子不足BTD单元及其衍生物。另一方面在M9和M10可逆的结果观察到的轨道电子的电荷重心和孔没有表现出在分子而不是单独的片段在分子轨道共享。激发后,电子密度转向BDTD BTD和DFBTD-from CDD情节,孔密度对BDTD单位。而对于M11公路和M12电子和空穴密度DCNBTD展出,DNBTD和BDTD单位。片段的贡献在分子轨道能级结果支持上述结果(图5 b)。电荷转移的长度(? r)是基于轨道参与电子的电荷重心的转变。电荷转移特征长度短(之间的分子?r = 1.5 (A)——激发/价激励)和长期(?励磁的r = 2.0)。TDDFT增加间距离计算的方法表明,电子交换的比例的增加,增加的电子转移的距离。从四个单体的转换系统M1只有间距离低于2.0 (图5一个)将只在分子本身价转换,而剩下的所有系统? r = 2.0。结果11描述系统(M2-M12)倾向于捐赠系统的电子从一个地方转移到另一个地方的受体系统。从上面的结果DTBT和DTBTD捐赠者字符而不是受体和M9-M12四个单体系统模拟光电属性显示选择双极性半导体。

图5:)研究分子的电荷转移距离在电子跃迁和b。碎片贡献在十二个分子的分子轨道能级(H-represents-HOMO;L-represents-LUMO)。
EWG开路电压是如何影响的?
开路电压(Voc)光伏电池直接相关共轭系统的功率转换效率与短路电流(Isc),直接取决于系统的带隙,这些因素决定形式填充因数的大小决定能源光伏电池的转换效率。开路电压Voc表示为:
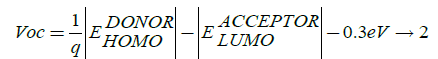
在这里,q是元电荷;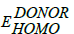 是捐赠的能级;
是捐赠的能级;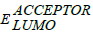 是受主能级和0.3 eV是一个经验因素。在理论方法开放电路可以由1 / q (q是单位电荷)乘以最高占据分子轨道的能级捐赠者- 4.3 V的LUMO能级的电脑60BM元电荷为0.3 V是一个经验性因素之间的能量差LUMO的供体和受体的LUMO。在亲水界面所需最小能量为0.3 V攻击激子分离和电荷分离。据汤普森等人报道的理想供体共轭聚合物系统应该3.9 eV LUMO和5.4 eV的人类;这样我们可以缓解逃避浪费的能量激子分离(70年]。高开路电压(Voc)造成低洼的HOMO能级的共轭体系。这样可以缓解平衡孔和供体和受体之间的电子迁移率聚合物系统(71年]。基于无机材料的硅,CdTc太阳能电池的开路电压(Voc)或多或少等于一个光学带隙(如)72年]。相反,在一个有机共轭体系开路电压低于光学带隙产生难以分离的激子数字-模拟接口。我们已经成功地确定了开路电压(Voc)的十二个单体一起电脑61年BM和ITIC-Th1受体。理论上预测开路电压的分子都是表1中讨论,表明大部分的单体生产5到-5.9 eV的HOMO能级。因此,人类稳定能量产生的派生高开路电压方程1。开路电压逐渐增加的顺序M12 > M11公路> M10 > M8 > M7 > M9 > M4 > M3 > M6 > M5 > M2 > M1。在这种背景下,我们报告单体与非常低,HOMO能级产生最高的Voc。有趣的是,七个分子具有高挥发性有机化合物> 0.80 V在所有,这是对小分子明显增强。特别是M12的开路电压、M11公路和M10保留Voc > 1由于地势低洼的HOMO能级。电子(EWG)退出组织培养Voc水平发挥了重要作用,而未被取代的衍生物巨大的差异被观察到0.40 - -0.46 V。高开路电压1.59 V时观察到ITIC-Th1用作n受体分子设计。最好的结果是发现分子M8低带隙的1.85 eV高开路电压为1.26 V时ITIC-Th1用作受体。 This above result showed EWG is unavoidable substituents should consider for designing Donor-Acceptor architectures to achieve high power conversion efficiency solar cells.
是受主能级和0.3 eV是一个经验因素。在理论方法开放电路可以由1 / q (q是单位电荷)乘以最高占据分子轨道的能级捐赠者- 4.3 V的LUMO能级的电脑60BM元电荷为0.3 V是一个经验性因素之间的能量差LUMO的供体和受体的LUMO。在亲水界面所需最小能量为0.3 V攻击激子分离和电荷分离。据汤普森等人报道的理想供体共轭聚合物系统应该3.9 eV LUMO和5.4 eV的人类;这样我们可以缓解逃避浪费的能量激子分离(70年]。高开路电压(Voc)造成低洼的HOMO能级的共轭体系。这样可以缓解平衡孔和供体和受体之间的电子迁移率聚合物系统(71年]。基于无机材料的硅,CdTc太阳能电池的开路电压(Voc)或多或少等于一个光学带隙(如)72年]。相反,在一个有机共轭体系开路电压低于光学带隙产生难以分离的激子数字-模拟接口。我们已经成功地确定了开路电压(Voc)的十二个单体一起电脑61年BM和ITIC-Th1受体。理论上预测开路电压的分子都是表1中讨论,表明大部分的单体生产5到-5.9 eV的HOMO能级。因此,人类稳定能量产生的派生高开路电压方程1。开路电压逐渐增加的顺序M12 > M11公路> M10 > M8 > M7 > M9 > M4 > M3 > M6 > M5 > M2 > M1。在这种背景下,我们报告单体与非常低,HOMO能级产生最高的Voc。有趣的是,七个分子具有高挥发性有机化合物> 0.80 V在所有,这是对小分子明显增强。特别是M12的开路电压、M11公路和M10保留Voc > 1由于地势低洼的HOMO能级。电子(EWG)退出组织培养Voc水平发挥了重要作用,而未被取代的衍生物巨大的差异被观察到0.40 - -0.46 V。高开路电压1.59 V时观察到ITIC-Th1用作n受体分子设计。最好的结果是发现分子M8低带隙的1.85 eV高开路电压为1.26 V时ITIC-Th1用作受体。 This above result showed EWG is unavoidable substituents should consider for designing Donor-Acceptor architectures to achieve high power conversion efficiency solar cells.
预测太阳能电池特性
Scharber图和他的同事设计了一个叫做“Scharber图”,这张图构造基于捐赠者的LUMO水平和带隙聚合物系统与PC60BM受主能级。他们报告说,在某种程度上使用“Scharber图”预测的功率转换效率设计共轭小分子和聚合物理论上可以提供精确的结果与实验结果和兼容性(11,73年- - - - - -75年]。“Scharber”图是广泛用于评估散装异质结太阳能电池的能量转换效率理论上混合标准PCBM受体。在这项研究中,我们使用fullerene-PC61BM和non-fullerene-ITIC-Th1受体接近结构的预测PCE。的富勒烯受体PC61BM广泛众所周知标准受体由于良好的溶解性,高的电子迁移率,商业可用性和人类,LUMO能级分别为-6.1和-4.3 eV。这些能量水平足以把一个电子从足够的捐赠共轭聚合物体系。有机共轭聚合物的能量变换带隙的范围应该在1.2 - -1.9 eV。高PCE里程碑不能实现只有一个带隙,应该有广泛的吸收范围和高开路电压驱动的HOMO能级-5.2到-5.8,LUMO能级-3.7到-4.0 V给能干的电荷分离而使大的开路电压。在我们的研究表明,有九个分子M3, M4和M6-M12达到低洼最高占据分子轨道能级的< -5.2 eV。三个单体M8、M11公路和M12最低能级之间的-3.7到-4.0 eV。所示图6的功率转换效率研究了三类结构分区2 - 5%,5 - 7%和M6 7 - 8%, M10;M3, M4 M7和M9;和M8。在所有分子M8显示更好的结果。

图6:近似的能量转化效率研究小分子利用Scharber预测图。
通过考虑p-extended共轭系统供体和受体之间插入单元伴随着H的替代高电负性的电子撤回(原子/组)取代基如F, CN和NO2在benzothiadiazole捐赠单位产生的先后十二单体设计。九个分子完全平面配置12个系统。电子捐赠趋势的防晒霜,DT-BTD和电子捐赠受体和捐赠者BDTD的性格截然不同的情况证明了电荷密度差异,电子空穴分布、电荷转移长度、分子轨道空间分布和连同片段分子能级的贡献。片段分子能量的贡献是重要的研究证明DTBT捐赠者字符,DTBTD BDTD和双重性格。功率转换效率的新设计共轭单体由Scharber方法;从理论上预测最大PCE单体M8在所有的7 - 8%。上述理解从理论调查获得小分子/单体提供深刻理解设计和调优能级遵守光学性质的共轭聚合物共轭材料光电设备。
没有冲突要申报的东西。
这项工作由科技部支持(No-CS-069/2013),印度政府。SC谢谢,信息通信技术部门仙露Vishwa Vidyapeetham提供计算工具。
